This thesis focuses on chorismate mutase (CM), an enzyme involved in the biosynthesis of aromatic amino acids in bacteria, fungi and plants. Novel exported bifunctional fusion enzymes with chorismate mutase and cyclohexadienyl dehydratase activity: shikimate pathway enzymes interact in no man's land.
Understanding bacterial metabolic pathways
Once a potential drug target is identified, its structure can be determined and used in structure-based drug discovery (Greer et al. 1994, Lionta et al. 2014, Batool et al. 2019). Structural determinants of potential binding pockets serve as the basis for identifying efficient ligands (Greer et al.
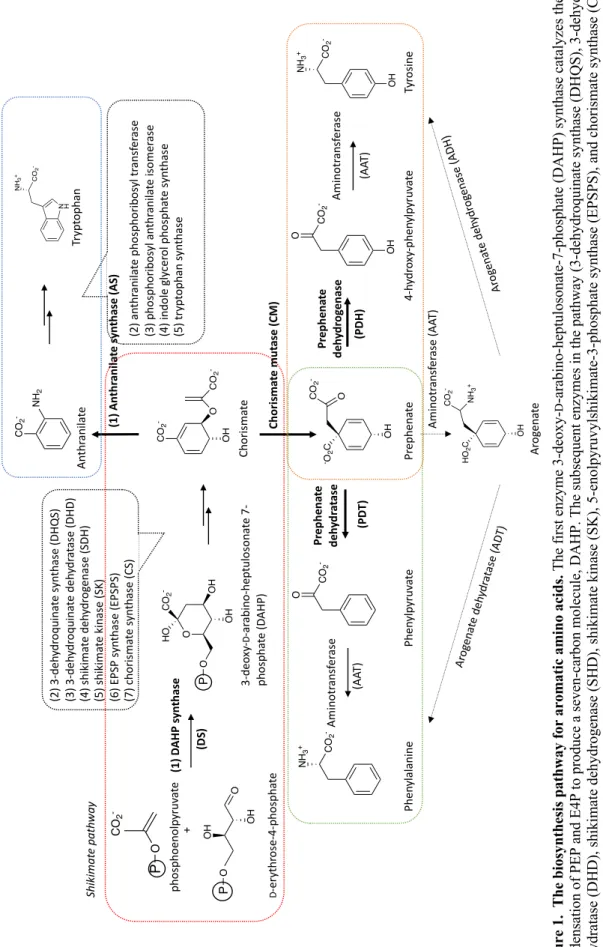
Aromatic amino acid biosynthesis pathway
- Shikimate pathway- common route to the biosynthesis of the aromatic amino acids
- Post-chorismate routes to the biosynthesis of aromatic amino acids
- Subcellular localization of the aromatic amino acid biosynthesis pathway
- Regulatory points in the biosynthesis of aromatic amino
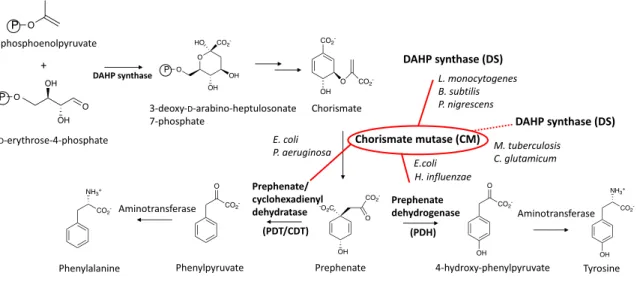
- DAHP synthase (DS)
- Chorismate mutase (CM)
- Pathogenic organisms depending on the aromatic amino acid biosynthesis pathway
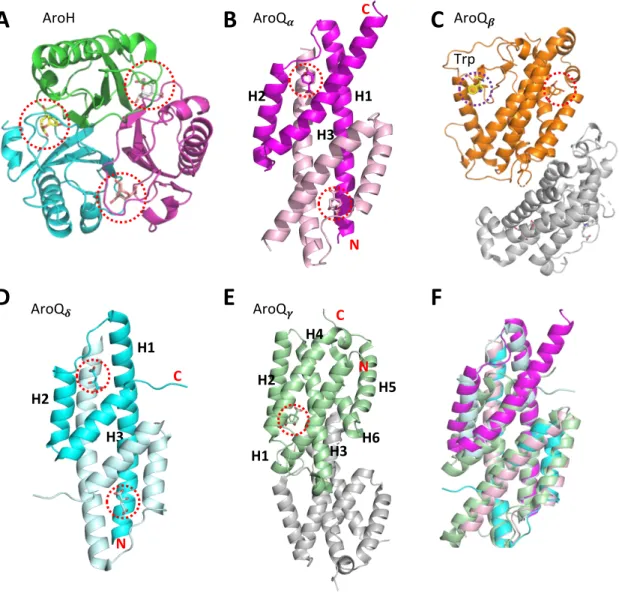
In bacteria, the biosynthetic pathway of aromatic amino acids is localized in the cytoplasm (Tzin et al. 2012). The AroQb subclass is represented by CM from Saccharomyces cerevisiae (ScCM) (Sträter et al. 1997) (Figure 4 C).
Enzyme colocalization
Substrate exchange mechanisms between colocalized enzymes
Electrostatic highway between active sites of bifunctional dihydrofolate reductase-thymidylate synthase (PDB ID: 1SEJ, (Svedružić et al. 2020)). A noncovalent heterooctamer complex of bifunctional CM-DS from Listeria monocytogenes (PDB ID: 3NVT, (Light et al. 2012)).

Model system: bifunctional extracytoplasmic *CDTCM and *CMCDT enzymes
Manuscript I, Summary
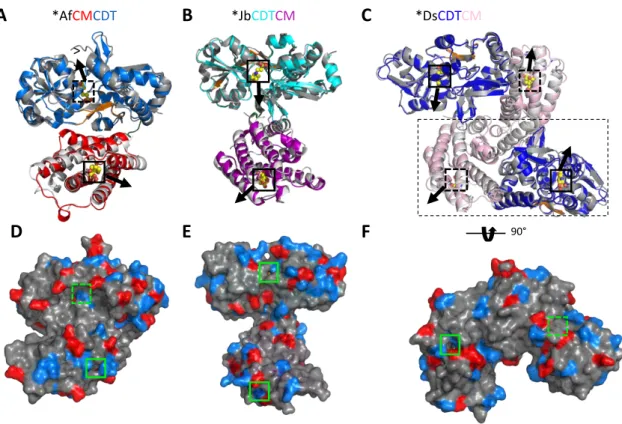
However, the opening of the active sites of the two domains points in opposite directions, making substrate channeling by nearby exchange unlikely (Figure 9 F). Overall, structural analysis of the fusion enzymes did not support the substrate channeling hypothesis.
Further insights from the crystal and solution structures of the bifunctional CM/CDTs
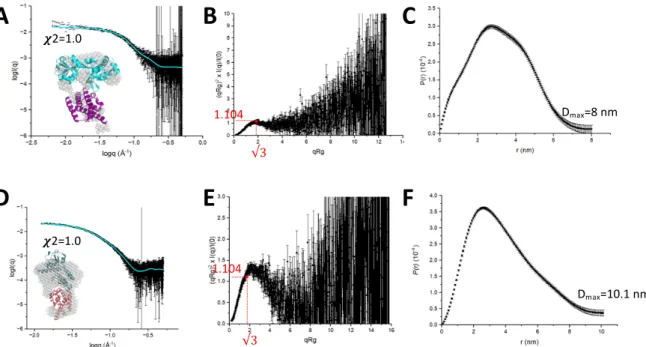
Fit between the experimental dispersion curve (black) and the theoretical (cyan) dispersion curve of *JbCDTCM calculated using Pepsi-SAXS from its crystal structure, with a c2 value of 1.0. Fit between the experimental (black) dispersion curve and the theoretical (cyan) dispersion curve of *ScCMCDT calculated using Pepsi-SAXS from its AlphaFold2 model with a c2 value of 1.0.

Model system: extracytoplasmic aromatic amino acid biosynthesis pathway enzymes from
Manuscript II, Summary
Fit between the experimental (black) scattering curve and the theoretical (cyan) scattering curve of *PaeCM calculated using Pepsi-SAXS from its dimer, with a c2 value of 1.1. Fit between the experimental (black) dispersion curve and the theoretical (cyan) dispersion curve of *PaeCDT calculated using Pepsi-SAXS from its trimer with a c2 value of 0.9.
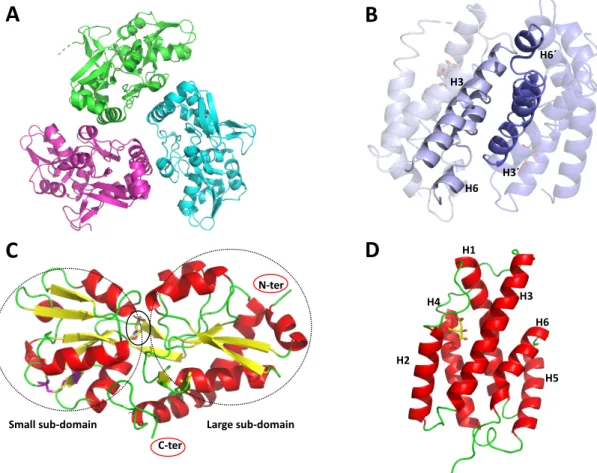
Additonal insights into CM and CDT domains
CDT domains
- Hinge-mediated open and closed conformation of the active site of CDT domain
- Intrinsically flexible region neighboring the b-strand (hinge region) of the CDT
- Extended C-terminus enabling interactions between the large and small subdomains 47
- Binding affinity for chorismate for the CM domains of extracytoplasmic CM/CDT
Conformations of the intrinsically flexible region of the CDT domain of the bifunctional enzymes observed in crystal structures. The C-terminus of the CDT domain of bifunctional enzymes shows the same conformation observed in *PaeCDT, binding the large subdomain by polar interactions. The CM domain of the bifunctional enzymes shows a tighter substrate gating compared to *MtCM (PDB ID: 2FP2, (Okvist et al. 2006)).
Normalized B-factor analysis (Johnson et al. 2018) of the bifunctional enzymes revealed that the H3-H4 loop is the most mobile region in *JbCDTCM and *AfCMCDT, followed by the. Normalized B-factor analysis of the crystal structures of CM domain of TSA-bound and apo-bifunctional enzymes compared to the monofunctional *MtCM. In Manuscript I, key residues in the active site of the CM domain of the *JbCDTCM,.
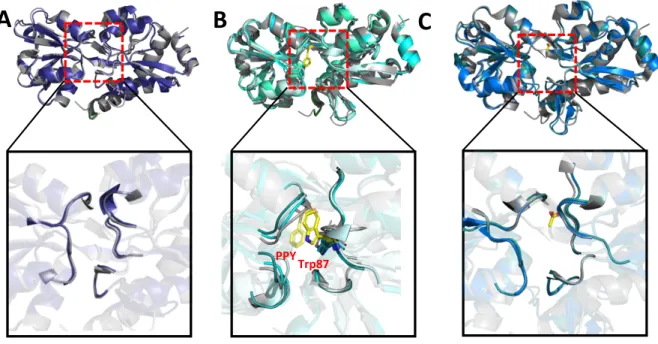
Model system: CM and DAHP synthase from Mycobacterium tuberculosis
CM-DS inter-enzyme allostery
Allosteric regulators, Phe, Tyr and Trp, bound to DS are depicted as sticks at the dimerization and tetramerization interfaces. The distance between Tyr and Phe binding sites and the nearest CM active site is shown as a dashed line (yellow). The interaction between CM and DS, which enables the interaction of the C-terminus of CM with a kinked H1-H2 loop.
Allosteric CM-DS feedback regulation by aromatic amino acids
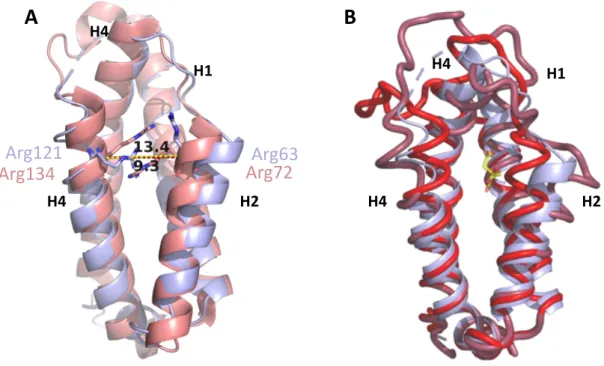
Superimposition of wild-type malate-bound MtCM (PDB ID: 2VKL, (Sasso et al. 2009); purple) and TSA-bound MtCM from the MtCM-MtDS complex (PDB ID: 2W1A, (Sasso et al. 2009) ; pink) , showing a significant conformational difference in the H1-H2 loop and the C-terminus. Two substitutions in the H1-H2 loop, four in the C-terminus, two in the N-terminal region, and three in the inter-subunit region. The same feature was observed in the crystal structure of a superactive variant of MtCMV (Fahrig-Kamarauskait et al. 2020), in contrast to the extended conformation observed in wild-type, low-activity MtCM (PDB ID: 2VKL, 2QBV (Kim et al. 2008, Sasso et al. 2009)) (Figure 28 B).
In active MtCM, the side chain of Arg46, a catalytically important residue, adopts a catalytically favorable conformation, whereas in wild-type apo MtCM this residue points away from the substrate and adopts a catalytically unfavorable conformation (Figure 28 C). Substitutions introduced in the H1-H2 loop, T52P, and V55D improved the kcat/Km of MtCM by 6-fold, 12-fold, and 22-fold, respectively, in combination. In manuscript III, we aimed to gain a deeper understanding of the functional significance of critical substitutions introduced in superactive MtCMV using enzyme kinetics, crystal structures, and molecular dynamics simulations.
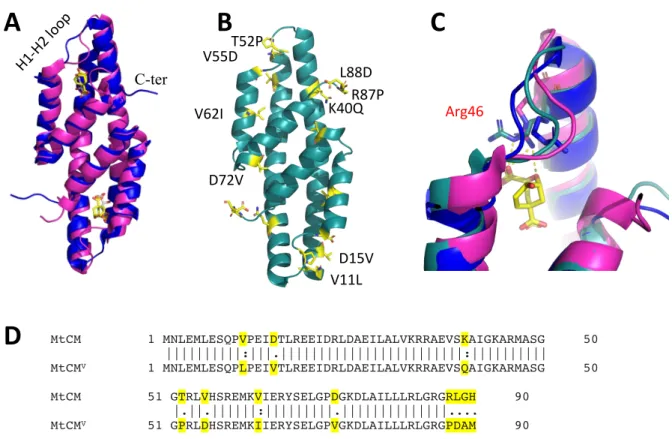
Manuscript III, Summary
Kinked H1-H2 loop: The conformational landscape of the H1-H2 loop was explored during the simulation using Arg53 as a reported residue. The H1-H2 loop in MtCMV and TSA-bound MtCMLC retained its kinked conformation during the simulation, whereas in wild-type, apo MtCM, it was found preferentially in extended conformation. Asp55 substitution allows salt bridging to Asp 46 and Arg18', promoting preorganization of the active site region (Figure 30).
The right panels show the distance between the Cζ carbon of arginine residue 53 or 46 and the carboxylate carbon of the C-terminus of MtCM in the two protomers (upper and lower panels). Formation of a stable contact (< 5 Å) with Arg46 (bottom panel) is consistent with stabilization of the catalytically productive H1-H2 loop conformation, allowing stabilization of the transition state of the chorismate to prephenate rearrangement. In MtCMV, a newly introduced Asp88 provides an alternative mode of salt bridging with Arg53, thus strengthening the H1-H2 loop and C-terminal interaction throughout the simulation.

Fragment-screening for MtCM (preliminary results from FragMAX and DSF)
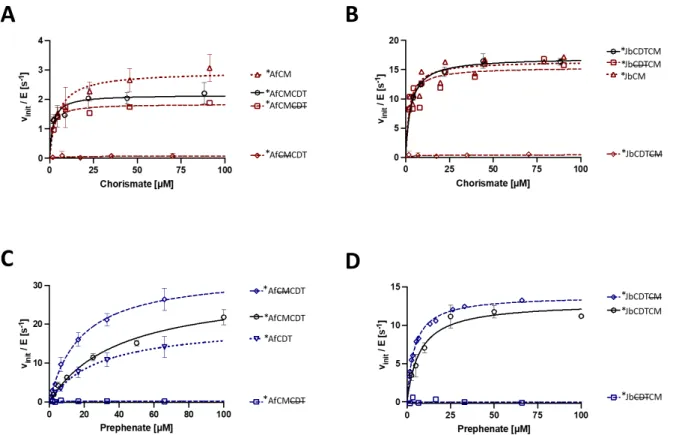
We investigated the presence of a catalytic advantage conferred by the fusion of the two domains using enzyme kinetics. The relative orientation of the active sites of CM and CDT and the existence of substrate channelization was investigated by X-ray crystallography. However, we did gain insight into the dynamic behavior of the CDT and the AroQγ subclass CM domains.
In the case of the latter, previous observations revealed the existence of an open and a closed state for the catalytic cleft, a feature confirmed by the structural analysis on the CM domain of bifunctional enzymes. AroQδ subclass CMs undergo inter-enzyme allosteric activation by forming a complex with DAHP synthase (DS), the first enzyme in the aromatic amino acid biosynthetic pathway. Further simulations of the full CM-DS complex may provide further understanding of the inter-enzyme allosteric process.

Recombinant gene expression and protein purification
Novel and exported bifunctional *CMCDT and *CDTCMs
Untagged and His 6 -tagged *PaeCDT and *PaeCM
20 mM BTP, pH 7.5, 150 mM NaCl 20 mM BTP, pH 7.5, 150 mM NaCl 20 mM BTP, 150 mM NaCl pH 7.5 Personally Purified Yes Yes Yes Yes AC: Affinity Chromatography AEX: Anion Exchange Chromatography CEX: Cation Exchange chromatography SEC: size exclusion chromatography.

Mycobacterium tuberculosis CM wild type expression and purification
Differential scanning fluorimetry (DSF)
- Introduction to the method
- Experimental procedure
- Mycobacterium tuberculosis chorismate mutase
- Bifunctional *CDTCM and *CMCDT
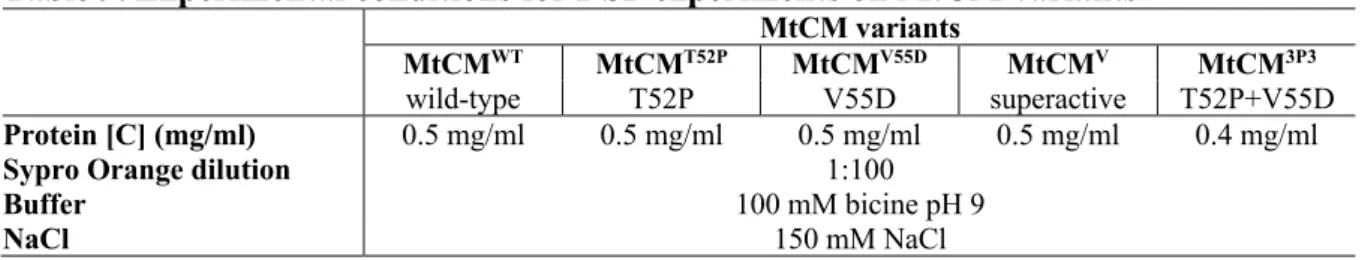
The melting temperature was calculated by non-linearly fitting the data with a sigmoid Boltzmann function through GraphPad Prism software, version 5 (GraphPad Software Inc., La Jolla, CA, USA). Prior to the actual stability experiments, preliminary screenings were performed to establish the optimal experimental conditions for all targets, using different combinations of protein concentrations (0.05–0.5 mg/ml) and Sypro Orange dilutions (Sigma Aldrich, 100-1000-fold diluted). Thermal stability of untagged MtCM variants was assessed by differential scanning fluorimetry using a protein concentration of 0.5 mg/ml (wild type: MtCMWT, T52P and V55D single mutants: MtCMT52P and MtCMV55D, super active mutant MtCMV52P 4 mg/ml ) or Mt. /ml (T52P+V55D double mutant: MtCMDP) in the presence of Sypro Orange diluted to a final volume ratio of 1:100, 100 mM bicine pH 9, 150 mM NaCl.
The best buffer for DSF experiments on bifunctional *CDTCM and *CMCDT variants was selected by pre-screening buffers from the Rubic Additive kit (Molecular Dimensions Ltd.). The buffer pre-screening experiment was performed using 0.5 mg/ml bifunctional proteins resuspended in their storage buffer and Sypro Orange diluted to a final volume ratio of 1:200. Subsequent protein stability experiments were performed at a final protein concentration of 0.5 mg/ml in a 100 mM Tris-HCl, pH 8.5, 150 mM NaCl buffer, 1:200 Sypro Orange dilution.
Protein X-ray crystallography
- Overview of the method
- Protein crystallization
- Introduction to the method
- Crystallization experiments
- Ligand-bound complexes crystallization
- Data collection
- Data processing and the structure determination
Protein crystallization can be illustrated by the phase diagram for a simplified system formed by a solution of the protein and the precipitant (Figure 33). The volume of the crystallization droplets varied between 400 and 600 nL; protein stocks were mixed at a volume ratio of 1:1 or 1:2 with crystallization solutions. In the latter case, the ligand was either dissolved in the mother liquor or added to it in solid form.
To prevent diffusion of the ligand from the crystal during cryoprotection, ligands were also added to the cryoprotectant droplets. Search patterns were found by performing a BLASTP search of target protein sequences restricted to proteins with a single PDB entry ( Johnson et al. 2008 ). As a final step for all structures, occupancy refinement was performed with phenix.refine, a tool of the PHENIX software package (Afonine et al. 2012).
Fragment-based screening
Introduction to the method
Fragment-based screening using X-ray crystallography
- Crystallization and soaking
- Data collection and processing
Ideally, crystals for screening fragments should be robust, large in size (>100 μm) (Collins et al. 2018), with diffraction better than 2 Å resolution and, most importantly, easily reproducible (Chilingaryan et al. 2012, Collins et al. 2018). The general workflow of X-ray crystallography-based fragment screening includes crystallization of the target protein, preparation of a protein-fragment co-crystal (either by soaking or co-crystallization), data collection, data processing, and analysis of bound ligands (Patel et al. 2014, Lima et al. 2020). For screening, we used FragMAXlib, a library developed at the FragMAX facility (MAX IV synchrotron, Lund, Sweden) and consisting of 171 fragments (Lima et al. 2020).
All datasets were processed by multiple automated pipelines in the FragMAX facility (XDSAPP, xia2/DIALS, xia2/XDS, fastdp, EDNA_proc) (Lima et al. 2021). After bin processing, PanDDA (Pan-Dataset Density Analysis) software (Pearce et al. 2017) was used to detect potential binding events. Event maps identify regions that are significantly different from the baseline condition (Pearce et al. 2017).

Orthogonal validation using thermal shift assay
- Experimental work
Data collection took place at cryogenic temperature (100K) at the BioMAX beamline, MAX-IV synchrotron light source (Lund, Sweden). The software identifies 'crystallographic events', such as a binding ligand, by subtracting the average of dozens of ground-state ("unbound") datasets from a dataset with an altered state ("bound"); this produces a residual partial difference map, called an event map. Samples were prepared from 1M stock solutions of the fragments dissolved in pure ethylene glycol, resulting in a final concentration of 4% v/v ethylene glycol.
Sypro Orange was diluted in water from a stock solution of the dye stored in pure DMSO. The melting temperature of native MtCM was calculated from five independent measurements, while for the fragments a single measurement was made. Diffraction limits and eigenvalues of overall anisotropy tensor on |F|s are displayed along the corresponding principal axes of the ellipsoid fitted to the diffraction cutoff surface as direction cosines in the orthogonal basis and in terms of reciprocal unit cell vectors.

Small-angle X-ray scattering
Principles of the method
Sample preparation
Data collection and processing
34;Crystal structures of a monofunctional chorismate mutase from Bacillus subtilis and its complex with a transition state analog." Proc. 34;Use of three-dimensional structures of protein target molecules in structure-based drug design." J. 34; Correct phylogenetic relationship of KdsA (3-deoxy-D-manno-octulosonate 8-phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-D-arabino-heptulosonate 7-phosphate synthase). "J.
34;A comparative biochemical and structural analysis of the intracellular chorismate mutase (Rv0948c) of Mycobacterium tuberculosis H(37)R(v) and the secreted chorismate mutase (y2828) of Yersinia pestis." FEBS J.